We spend our lives surrounded by hi-tech materials and chemicals that make our batteries, solar cells and mobile phones work. But developing new technologies requires time-consuming, expensive and even dangerous experiments.
Luckily we now have a secret weapon that allows us to save time, money and risk by avoiding some of these experiments: computers.
Thanks to Moore’s law and a number of developments in physics, chemistry, computer science and mathematics over the past 50 years (leading to Nobel Prizes in Chemistry in 1998 and 2013) we can now carry out many experiments entirely on computers using modelling.
This lets us test chemicals, drugs and hi-tech materials on a computer before ever making them in a lab, which saves time and money and reduces risks. But to dispense with labs entirely we need computer models that will reliably give us the right answers. That’s a difficult task.
A grand challenge
Why so difficult? Because chemistry is the quantum mechanics of interacting electrons – usually based on Schrodinger’s equation – which require enormous amounts of memory and time to model.
For example, to study the interaction of three water molecules, we need to store around 1080 pieces of data, and do at least 10320 mathematical operations.
This basically means that when the universe ends we’d still be waiting for an answer. This is somewhat of a bottleneck.
But this bottleneck was broken by three major advances that allow modern computer models to approximate reality pretty well without taking billions of years.
Firstly, Pierre Hohenberg, Walter Kohn and Lu Jeu Sham turned the interaction problem on its head in the 1960s, greatly simplifying and improving theory.
They showed that the electronic density – a quantum mechanical probability that is fairly easy to calculate – is all you need to determine all properties of any quantum system.
This is a truly remarkable result. In the case of three water molecules, their approach needs only 3,000 pieces of data and around 100 billion maths operations.
Secondly, in the 1970s John Pople and co-workers found a very clever way to simplify the computing method by employing mathematical and computational shortcuts.
This lets us use just 300 pieces of data for three water molecules. Calculations need around 100 million operations, which would take a 1975 supercomputer two seconds but can be solved 500 times in a second on a modern phone.
And finally, the 1990s saw a bunch of people come up with some simple methods to approximate very complex interaction physics with surprisingly high accuracy.
Modern computer models are now mostly fast and mostly accurate, most of the time, for most chemistry.
Quantum mechanical modelling takes off
As a result, computer modelling has transformed chemistry. A quick glance through any recent chemistry journal shows that many experimental papers now include results from modelling.
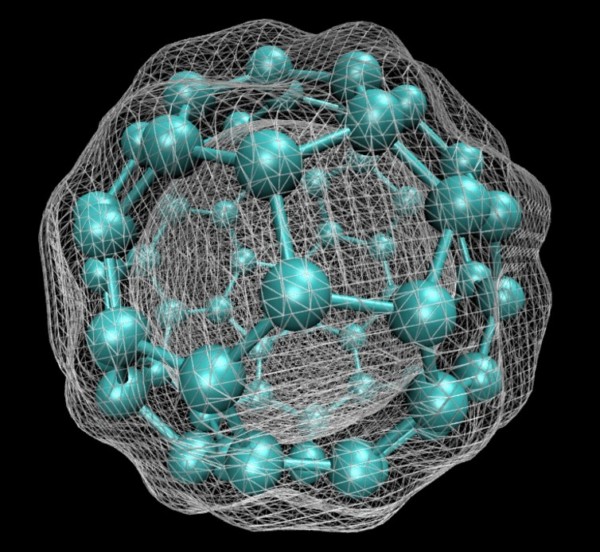 Computers can show quantum mechanical details no experiment can probe. Here modeling has been used to calculate and plot the electron density of a C60 bucky ball. Itamblyn/Wikipedia
Computers can show quantum mechanical details no experiment can probe. Here modeling has been used to calculate and plot the electron density of a C60 bucky ball. Itamblyn/Wikipedia
Density functional theory (the technical name for the most common modelling method) is a feature in more than 15,000 scientific papers published in 2015. Its impact will only continue to grow as computers and theory improve.
Modelling is now used to uncover chemical mechanisms, to reveal details about systems that are hidden from experiments, and to propose novel materials that can later be made in a lab.
In a particularly exciting case, computers were able to predict that a molecule C3H+ (propynylidynium) was responsible for some strange astronomical observations.
C3H+ had never before been seen on Earth. When it was later made in a lab it behaved just as the modelling predicted.
New challenges need new solutions
However, the rise of graphene exposed a major flaw in existing models.
Graphene and similar 2D materials do not stick together in the same way as most chemicals. They are instead held together by what are known as van der Waals forces that are not included in standard models, making them fail in 2D systems.
This failure has led to a surge of interest in computer modelling of van der Waals forces.
For example, I was involved in an international project that used sophisticated modelling to determine the energy gained by forming graphite out of layers of graphene. This energy still cannot be determined by experiments.
Even more usefully, 2D materials can potentially be stacked like LEGO, offering vast technological promise. But there are basically an infinite number of ways to arrange these stacks.
We recently developed a fast and reliable model so that a computer can churn through different arrangements very quickly to find the best stacks for a given purpose. This would be impossible in a real lab.
On another front, electrical charge transfer in solar cells is also difficult to study with existing techniques, making the models unreliable for an important field of green technology.
Even worse, highly promising (but dangerous) lead based perovskite solar cells involve van der Waals forces and charge transfer together, as shown by some colleagues and me.
A substantial effort is underway to deal with this difficult problem, and the equally difficult (and related) magnetism and conduction problems.
Things will only get better
The ultimate goal of computer modelling is to replace experiments almost entirely. We can then build experiments on a computer in the same way people build things in Minecraft.
The computer would model the real world to allow us to save real time and money and avoid real dangerous experiments.
For example, the Titan supercomputer (pictured top) has recently been used to study non-icing surface materials at the molecular level to improve the efficiency of wind power turbines in cold climates.
This ultimate goal was almost met in the 1990s until the experimental scientists came up with graphene and perovskites that showed flaws in existing theories. Researchers like me continue to study, anticipate and fix these flaws so that computers can replace more challenging experiments.
Perhaps the 2020s will be the last decade when experiments are carried out before knowing what the answer will be. That is a certainly a model worth striving for.![]()
By Timothy Gould, Lecturer in Physics, Griffith University. This article was originally published on The Conversation. Read the original article.


